Genomic signatures of selection

Species evolve constantly over time as individuals are born, reproduce and die. Doing so their genomes pass on from parents to offspring, modified by mutations and shuffled through recombination.
Constraints placed on the ability of individuals to survive in their environment, to chose their mates, to produce surviving offspring … will impact the way genomes change over time.
In return, patterns of diversity observed in present day genomes carry information on these constraints and therefore on the evolutionary history of populations in the past and on biological processes that affect the way genomes are transmitted.
My work in population genomics aims at developping new ways to reconstruct how selection has affected the diversity observed in the genome of (mostly) livestock species. So we try, via modeling, to interpret genetic data recorded on whole genomes in order to gain insight into the past history of populations.
Population differentiation
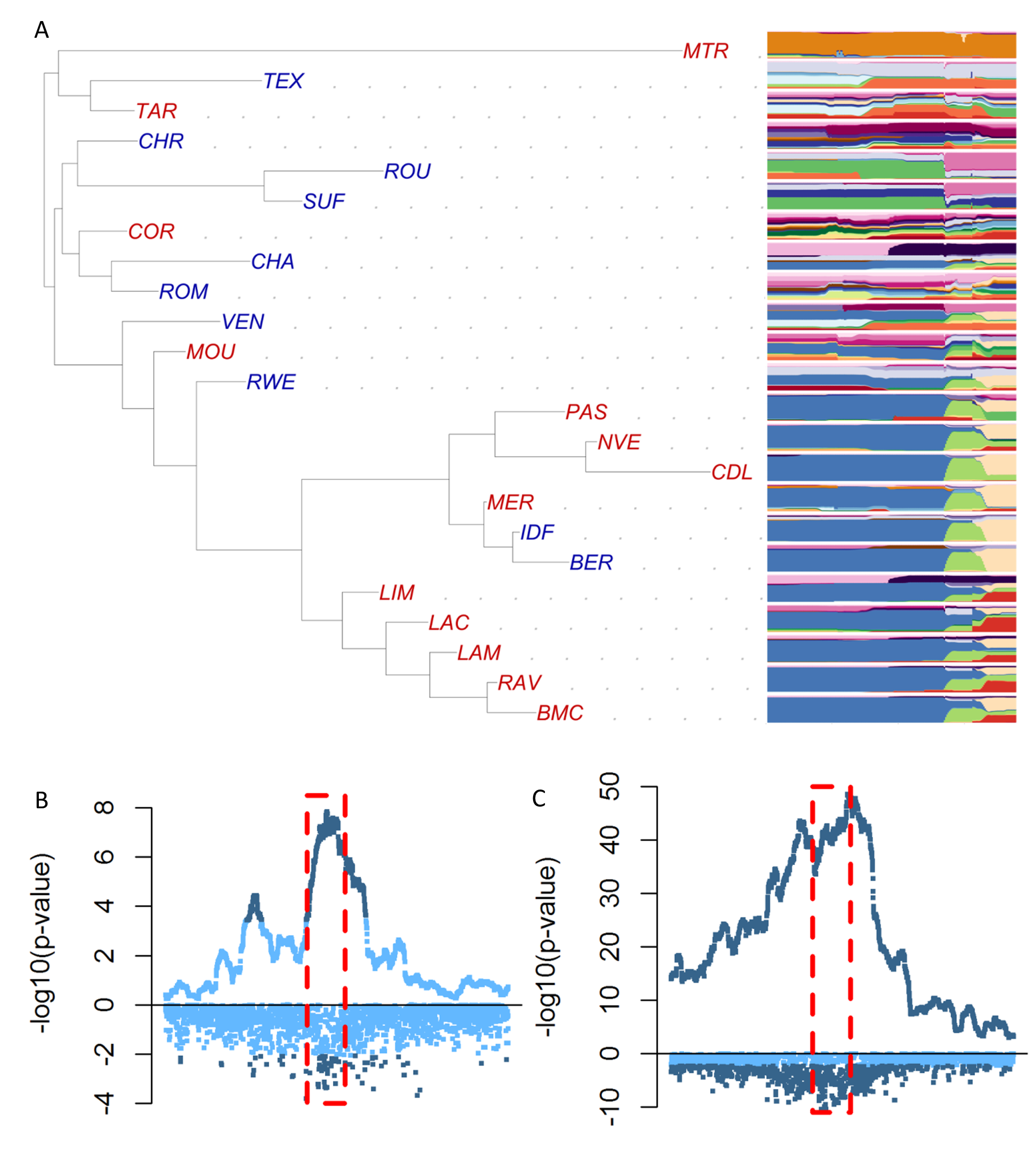
As illustrated above, one way to identify genomic regions under selection is to look for genome regions where populations carry very different genetic backgrounds, more than what we expect from their average, genome-wide, patterns. Building upon the work of Maxime Bonhomme (Bonhomme et al. 2010), we extended this approach to work on haplotypes rather than point mutations (Fariello et al. 2013) which increased interpretability and statistical power. Later on, we further worked to look specifically for signatures associated to selection on particular phenotypes of local pig breeds from Europe (Poklukar et al. 2023). These methods, including Maxime’s FLK, are implemented in the hapFLK software. We are currently working on extending the hapFLK method to improve the statistical inference on dating past selection events.
Genetic Time series
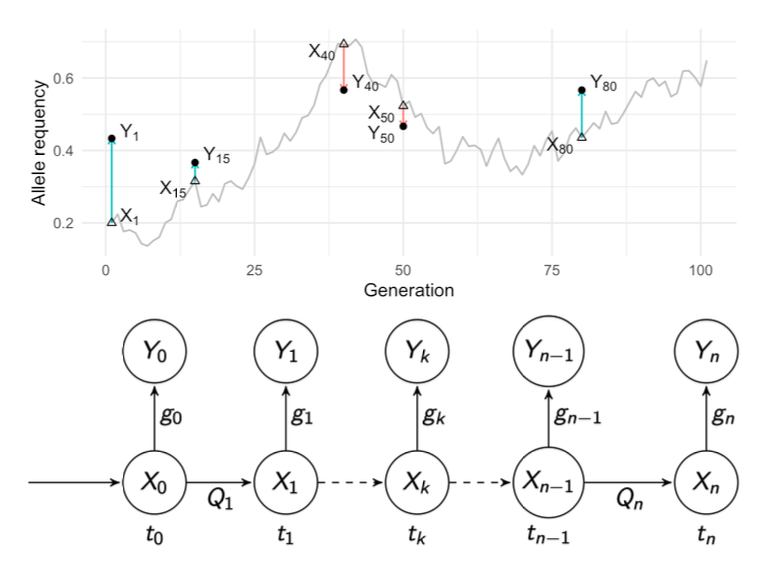
With the increased availability of dense genetic data, a new kind of information can be taken into account: time. From ancient DNA studies, experimental evolution experiments or continuous genetic survey of natural and domestic populations, we now have access to the evolution of the genetic composition of groups of individuals over time, genetic time series.
We have worked on one particular class of statistical models to analyse such data: Hidden Markov Models (HMM). The objectives of the analyses are to estimate for each allele along the genome a selection coefficient that explains the trajectory of its frequency over time. In (Paris, Servin, and Boitard 2019), we compared different models to approximate the transition kernels in such HMM. We found that the Beta-with-Spikes approximation was performing best. Since then, we have been working on using the HMMs to improve inference both on the effective population size and selection coefficients. These methods are implemented in the SelNeTime software (Uhl et al. 2025).